Review Article
Rhabdomyoblasts in Pediatric Tumors: A Review with Emphasis on their Diagnostic Utility
Giuseppe Angelico*, Eliana Piombino, Giuseppe Broggi, Fabio Motta and Saveria Spadola
Department of Medical and Surgical Sciences and Advanced Technologies, G.F. Ingrassia, Azienda Ospedaliero-Universitaria “Policlinico-Vittorio Emanuele”, Anatomic Pathology Section, School of Medicine, University of Catania, Catania, Italy
*Address for Correspondence: Giuseppe Angelico, Department of Medical and Surgical Sciences and Advanced Technologies, G.F. Ingrassia, Azienda Ospedaliero-Universitaria “Policlinico-Vittorio Emanuele”, Anatomic Pathology Section, School of Medicine, University of Catania, Italy, Email: [email protected]
Dates: Submitted: 23 December 2016; Approved: 07 March 2017; Published: 09 March 2017
How to cite this article: Angelico G, Piombino E, Broggi G, Motta F, Spadola S. Rhabdomyoblasts in Pediatric Tumors: A Review with Emphasis on their Diagnostic Utility. J Stem Cell Ther Transplant. 2017; 1: 008-016. DOI: 10.29328/journal.jsctt.1001002
Copyright License: © 2017 Angelico G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Rhabdomyosarcoma; Nephroblastoma; Malignant triton tumor; Pleuropulmonary blastoma; Rhabdomyoblast
SUMMARY
Rhabdomyosarcoma is a soft tissue pediatric sarcoma composed of cells which show morphological, immunohistochemical and ultrastructural evidence of skeletal muscle differentiation. To date four major subtypes have been recognized: embryonal, alveolar, spindle cell/sclerosing and pleomorphic. All these subtypes are defined, at least in part, by the presence of rhabdomyoblasts, i.e. cells with variable shape, densely eosinophilic cytoplasm with occasional cytoplasmic cross-striations and eccentric round nuclei. It must be remembered, however, that several benign and malignant pediatric tumours other than rhabdomyosarcoma may exhibit rhabdomyoblaststic and skeletal muscle differentiation. This review focuses on the most common malignant pediatric neoplasm that may exhibit rhabdomyoblastic differentiation, with an emphasis on the most important clinicopathological and differential diagnostic considerations.
INTRODUCTION
Rhabdomyosarcoma (RMS) is a relatively common soft tissue sarcoma that frequently affects children and adolescents, defined by morphological, immunohistochemical and ultrastructural evidence of skeletal muscle differentiation. To date four major subtypes have been recognized: embryonal, alveolar, spindle cell/sclerosing and pleomorphic [1]. All these subtypes are defined, at least in part, by the presence of rhabdomyoblasts, i.e. cells with variable shape, densely eosinophilic cytoplasm with occasional cytoplasmic cross-striations and eccentric round nuclei [2]. Rhabdomyoblasts are primitive mesenchymal cells showing variable degrees of differentiation towards skeletal muscle. The detection of this cells on histological examination is considered one of the main parameters though not essential for the diagnosis of rhabdomyosarcoma. Rhabdomyoblasts assume a variety of histologic appearances depending on their stage of differentiation [3]. The most primitive forms are stellate cells with amphophilic cytoplasm and central oval nuclei. Gradually this cells acquire more cytoplasmic eosinophilia and show a variety of elongated shapes described as “tadpole”, “strap” and spider cells. In the end stage of differentiation, rhabdomyoblasts assume a bright eosinophilic cytoplasm with evident cross-striation. Multinucleated giant cells derived from the fusion of several myoblasts and myotube forms may also be seen. Immunohistochemically, rhabdomyoblastic differentiation can be identified with desmin, myogenin and MyoD1, which are currently considered the most reliable immunomarkers of skeletal muscle differentiation [2]. It must be remembered, however, that a heterologous rhabdomyoblastic component may occur in tumors with sarcomatous differentiation, tumors of neuroectodermal derivation, tumors with epithelial components and tumors with germ cell or sex cord elements. Among soft tissue sarcomas, rhabdomyoblastic elements are described not only in rhabdomyosarcoma but also in several sarcomas, especially those undergoing dedifferentiation, including chondrosarcoma, liposarcoma and malignant mesenchymoma [4]. The awareness that rhabdomyoblastic and skeletal muscle differentiation may occur in pediatric tumors other than rhabdomyosarcoma is crucial in order to avoid confusion with other neoplasms in the differential diagnosis. This review will focus on the most common malignant pediatric neoplasm that may exhibit rhabdomyoblastic differentiation, with an emphasis on the most important clinicopathological and differential diagnostic considerations.
Rhabdomyosarcoma
Rhabdomyosarcoma is a malignant tumor that frequently affects children and adolescents. The most common sites of occurrence are the head and neck region, the genitourinary tract, the limbs and the trunk [2]. Rhabdomyosarcoma is composed of neoplastic cells which show variable morphologic, immunohistochemical and ultrastructural evidence of skeletal muscle differentiation. Based on morphological, immunohistochemical and genetic features, four major subtypes have been recognized: embryonal, alveolar, spindle cell/sclerosing and pleomorphic [5]. Embryonal rhabdomyosarcoma (ERMS), the most common subtype, consists of a proliferation of round or spindle shaped undifferentiated mesenchymal cells admixed with a variable number of rhandomyoblasts alternating with zones of loose, myxoid, paucicellular stroma. Rhabdomyoblasts may become the predominant cell population following chemotherapy for rhabdomyosarcomas. The number of cells with rhabdomyoblastic features varies each case, and some possess a large amount of differentiated cells that closely resemble rhabdomyoma. Embryonal rhabdomyosarcomas arising beneath a mucosal surface (bladder, vagina, biliary tract, pharynx, conjunctiva, auditory canal) and growing as grape-like, nodular or polypoid excrescences, are labeled “botryoid rhabdomyosarcoma” [6]. Spindle cell rhabdomyosarcoma, most commonly arises in the head and neck and paratesticular soft tissues and shows a striking male predilection. This rare subtype of ERMS is composed almost exclusively of fascicles of elongated spindle cells arranged in whorls or in a fascicular/stroriform growth pattern [7]. Sclerosing rhabdomyosarcoma is a tumor closely related to the spindle cell subtype, characterized histologically by a prominent hyaline or pseudochondroid stroma [8]. Certain cases of spindle cell and sclerosing RMS, especially those occurring in the paratesticular region, display histological features of embryonal RMS, including the presence of rhabdomyoblasts. Spindle cell/ sclerosing RMS, is associated with a relatively better clinical outcome, when compared with other subtypes of RMS. However, recent studies identified a specific MYOD1 (L122R) mutations in a subset of sclerosing/spindle cell RMS, which correlate with a relatively aggressive clinical course [9]. Alveolar rhabdomyosarcoma is a distinct subtype of rhabdomyosarcoma associated with aggressive behavior. It is a highly cellular neoplasm composed of undifferentiated round cells admixed with variable number of rhabdomyoblasts and multinucleated giant cells with peripheral nuclei [2]. The classical histological growth pattern, referred as “alveolar” is characterized by nests of round cells separated by fibrous septa that tend to peripheral discohesion so that the cells appear to float in alveolar spaces. Pleomorphic rhabdomyosarcoma is a high-grade sarcoma with poor prognosis affecting almost exclusively adults. On histological examination is characterized by a pattern less architecture with no evidence of embryonal or alveolar component and consist of large, bizarre polygonal, round and spindle cells which show skeletal muscle differentiation [10]. Immunohistochemically, desmin, myogenin and MyoD1 are currently considered the most reliable markes, being positive, with a variable intensity and extension, in all the subtypes of rhabdomyosarcoma. In addition, an important feature for the diagnosis of the alveolar subtype, is its strong and diffuse nuclear myogenin expression, with up to 80-100% of tumor cells positive, unlike the patchy, heterogeneous pattern often seen with embryonal rhabdomyosarcoma, with few to 80% of tumor cells positive [11].
Infantile Rhabdomyofibrosarcoma
Infantile Rhabdomyofibrosarcoma (IRMFS) is an exceedingly rare pediatric sarcoma observed in children from 3 months to 3 years of age. This tumor shows overlapping features between spindle cell rhabdomyosarcoma and congenital/infantile fibrosarcoma [12]. It was first reported in 1993 by Lundgren et al. who described three cases initially diagnosed as infantile fibrosarcoma (IFS). These tumors, in contrast to the favorable clinical course of infantile fibrosarcoma, showed an aggressive clinical behavior in terms of distant metastasis and local recurrence [13]. Microscopically, IRMFS displays a variable number of rhabdomyoblastic cells scattered within a spindle cell proliferation with desmoplastic portions simulating infantile fibrosarcoma. The neoplastic cells show positive immunohistochemical stain for vimentin, smooth muscle actin, and desmin but no myoglobin, myoD1 or myogenin stain is observed. Since reports of rhabdomyofibrosarcoma cases are limited in the literature, the exact nature of this tumor s still not clear. However, although rare, IRMFS must be differentiated from IFS since the prognosis and treatment of these neoplasms is different.
Malignant Peripheral Nerve Sheath Tumors
Malignant peripheral nerve sheath tumors (MPNST) represent a rare group of soft tissue sarcomas with aggressive clinical behavior and high incidence of distant metastases. They often arise from peripheral nerves and show differentiation toward one of the cellular components of the nerve sheath (Schwann cells, fibroblasts, and perineurial cells) [14]. They can originate either de novo or from a preexisting benign nerve sheath tumor (usually neurofibroma) In addition, approximately 50% of these neoplasms arise in patients with neurofibromatosis type 1 syndrome (NF1). When associated with NF1, MPNSTs tend to present at a younger age (28-36 years) than the sporadic counterpart (40-44 years) [15]. Patients often present with a rapidly enlarging mass that may cause pain and/or neurological symptoms. The proximal portion of the upper and lower extremities and the trunk, are the most common locations of these tumors. These neoplasms lack of specific morphological, immunohistochemical and molecular features and display a variety of architectural patterns and cell morphology. On histological examination, some MPNSTs show areas of divergent mesenchymal or even epithelial differentiation. Neoplastic cells showing skeletal muscle differentiation, in the form of rhabdomyoblasts, represents the most common line of divergent differentiation encountered in MPNSTs [14]. Rhabdomyosarcomatous elements within MPNSTs were first described by Masson in patients with neurofibromatosis [16]. For these neoplasms Woodruff et al. proposed the descriptive term of “Triton Tumor” on the basis of the discovery that supernumerary limbs containing bone, neural and muscular elements were induced to grow on the backs of triton salamanders by transplantation of the sciatic nerve into the soft tissues of the back [17]. Malignant triton tumor (MTT) is a subtype of MPNST presenting rhabdomyoblastic differentiation. The majority of reported cases are described in patients with NF1 but the tumor may also occur in sporadic form. In the latter eventuality, other spindle cell sarcomas such as the fibrosarcoma, rhabdomyosarcoma, osteosarcoma, chondrosarcoma, and liposarcoma should be included in the differential diagnosis. On histological examination, MTTs are composed of a variable number of rhabdomyoblasts scattered throughout a stroma with the typical features of MPNSTs. Rhabdomyoblasts are usually relatively mature with abundant eosinophilic cytoplasm and show polygonal, spindled or bizarre shapes. This cells should be distinguished from benign skeletal muscle infiltrated by an otherwise typical tumor of peripheral nerves. Approximately 15% of MTTs show additional mesenchymal or epithelial areas, a phenomenon called “pluridirectional differentiation” [14]. Immunohistochemically, the morphologic suspicion of rhabdomyoblastic differentiation is confirmed with positive staining for skeletal muscle markers such as desmin, myoglobin, or muscle actin. MTT have a worse prognosis and an aggressive clinical behavior compared to other forms of MPNST. The 2year and 5year survival rates of MTTs are respectively of 15 % and 11%. For comparison, the 2 year and 5-year survival rates for MPNSTs, included those with heterologous elements are reported to be 57% and 39% respectively [17].
Malignant Ectomesenchymoma
Malignant ectomesenchymoma (MEM) is a rare multiphenotypic pediatric sarcoma consisting of neuroectodermal and mesenchymal neoplastic elements. This unusual and rapidly progressing sarcoma is believed to originate from the ectomesenchyme, which is the term used for neural crest tissue that shows mesenchymal differentiation during embryogenesis [18]. Approximately 64 cases of MEM have been so far reported in the literature, the majority of which have arisen in young infants with a marked male predominance. The most common sites of occurrence include retroperitoneum, abdomen and pelvic-perineal area followed by intracranial region, head and neck, extremities, and mediastinum [19]. Histologically, MEM displays areas of rhabdomyosarcoma, most frequently of embryonal phenotype, with evident rhabdomyoblastic differentiation. The admixed neuroectodermal component displays features of ganglioneuroma, neuroblastoma, or malignant peripheral nerve sheath tumor (MPNST). However, a variety of other mesenchymal or neuroectodermal elements have been reported in the literature. To date, due to the limited number of studies, the pathogenesis of MEM and its relationship with embryonal rhabdomyosarcoma and malignant peripheral nerve sheath tumor (MPNST) has not yet been elucidated. However, the latest reports suggest that MEM might represent a variant of embryonal rhabdomyosarcoma [19].
Nephroblastoma (Wilms Tumor)
Wilms tumor (WT) is a malignant pediatric embryonal neoplasm that occurs as a result of abnormal cellular differentiation during organogenesis and is thought to originate from nephrogenic blastemal cells. This neoplasm replicates the histology of developing kidneys and often shows divergent lines of differentiation [20]. WT is the most common genitourinary tumor of childhood affecting approximately 1 per 8000-10000 children with a peak incidence between 2 and 5 years of age. Both kidneys are equally affected, the incidence of synchronous or metachronous bilateral involvement being 5-10% [21]. Clinically, Wilms tumors present as an abdominal mass felt by a parent, associated in some cases with abdominal pain, hematuria and hypertension. Abdominal crisis and other symptoms related to traumatic rupture are also common. On gross examination, most WTs are solitary large masses measuring more than 5 cm in diameter with a median weight of 500 g. usually a fibrous pseudo-capsule demarcates the tumor from the adjacent renal parenchyma. The cut surface shows a grayish or yellowish, solid or firm mass depending on the percent of mature stromal elements presents, with foci of hemorrhage, necrosis. Areas of cystic degeneration are frequently seen, while rare cases are extensively cystic. Occasionally, polypoid protrusion of tumor into renal pelvis is seen [22]. This feature is associated with prominent skeletal muscle differentiation and raises problems of differential diagnosis with rhabdomyosarcoma. Histologically, most Wilms tumors exhibit triphasic histological components, consisting of blastema, epithelium and stroma, however biphasic and monophasic tumors are not uncommon. The blastematous areas consist of densely packed small round cells with scanty cytoplasm, dark staining nuclei and frequent mitotic figures. Blastemal cells display several growth patterns, the most frequent being diffuse, nodular and cord-like. The epithelial component is often composed of tubules and glomerular structures that closely recapitulate the normal stages of nephrogenesis (Figure 1A). Primitive rosette-like structures representing early tubular forms and cysts lined by primitive columnar or cuboidal cells are usually seen. The stromal component exhibits a variety of patterns of differentiation. Most of tumors are composed of a spindle cell component set in a myxoid background, resembling embryonal mesenchyme. Heterologous stromal elements including skeletal muscle, cartilage, bone, fat, neuroglia and mature ganglion cells are frequently observed in WT. Skeletal muscle in various stages of differentiation, including rhabdomyoblasts (Figure 1B, C) represents the most common divergent line of differentiation [23]. Rare cases of WT showing diffuse and predominant rhabdomyoblastic differentiation have been reported in the literature under the descriptive term of fetal rhabdomyomatous nephroblastoma [24]. This exceedingly rare variant of Wilms’ tumour shows little or no response to preoperative chemotherapy and is associated with a generally favourable outcome in most of all unilateral cases when treated by surgery. The majority of nephroblastomas carry a favorable outcome and an excellent prognosis with limited local growth and metastatic potential. The most important clinical and morphological criteria to establish the prognosis and the therapeutic strategies for patients with WT are as follows: (i) age; (ii) stage; (iii) histology [25]. The assessment of anatomic extent of the tumor (stage) represents the most important prognostic factor. Capsular invasion, surgical margins, involvement of renal sinus vessels, tumor implants, lymph node metastases, distant metastases, and bilaterality are the main parameters used to establish the stage. Age at diagnosis is another important prognostic indicator. WTs diagnosed in children under two year of age show a favorable outcome and a lower metastatic potential compared to those diagnosed over 2 years. On the basis of the absence or presence of anaplasia, Wilms tumors are divided in two categories: favorable and unfavorable histology. Anaplastic cells show a combination of nuclear enlargement, hypercromasia and multipolar mitotic figures. Anaplasia, is distinguished in focal and diffuse, based on its extent, and is found in about 5% of WTs. This feature is associated with a poor prognosis and increased risk of treatment failure, especially when is diffuse [26].
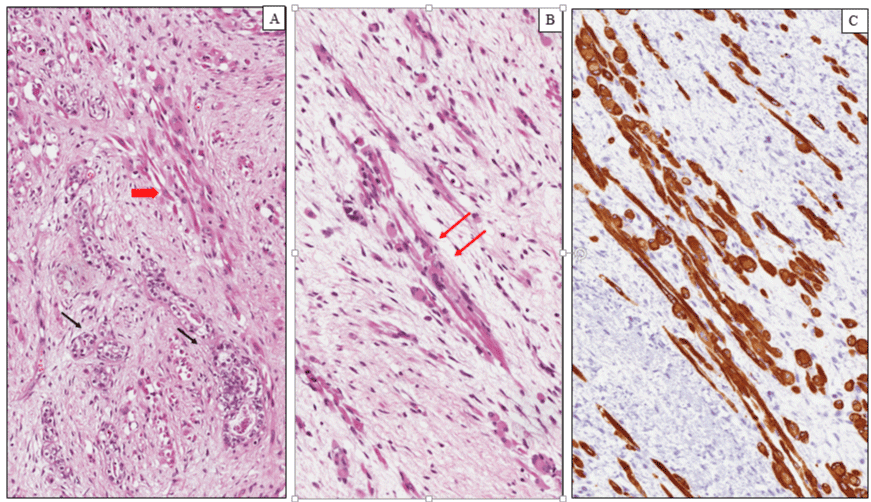
Figure 1: Nephroblastoma (Wilms tumor). (A) Histological examination showing a proliferation of primitive tubular ad glomerular-like structures (black arrows) admixed with rabdomyoblats of variable size and shape (red arrow). (B) Elongated and oval rhabdomyoblasts with eccentric round nuclei and densely eosinophilic cytoplasm with occasional cytoplasmic cross-striations (arrows). (C) Diffuse and intense cytoplasmic immunoreactivity for desmin in the rhabdomyoblastic component.
Pleuropulmonary Blastoma
Pleuropulmonary blastoma (PPB) is a malignant tumor of infancy and early childhood which belongs to the group of dysontogenic neoplasm such as Wilms tumor, hepatoblastoma, neuroblastoma, and embryonal rhabdomyosarcoma [27]. PPB occurs almost exclusively in children, younger than 5 years, as a pulmonary and/or pleural-based mass with cystic, solid, or combined cystic and solid features. There is evidence of a hereditary tumor predisposition syndrome in approximately 25% of cases [28]. The most common clinical manifestations include respiratory distress with or without pneumothorax, non-productive cough and fever, chest pain. Three main pathologic types of PPB have been described, each with different clinical presentation and prognostic features. Type I is purely cystic tumor, type II is mixed cystic and solid, type III is purely solid. Several studies suggest that type I PPB can progress to type II and type III PPB as the cysts are replaced by solid malignant tissue [29]. According to this model, the three types of PPB represent a continuum spectrum of lesions. The natural history suggests that many PPB begin as a lung cyst with a condensation of primitive mesenchymal element beneath the epithelium (type I). In subsequent stages, the mesenchymal cells expand and overgrow the cyst septa and replace the cyst with a cystic and solid (type II) or purely solid (type III) sarcomatous neoplasm. Not all type I PPBs progress to type II or III PPBs, and to date, no clinical or biological markers can predict which cysts will progress to sarcoma. Type I PPB is a purely cystic neoplasm with clinical and radiographic features indistinguishable from congenital lung cysts or congenital lobar emphysema. This neoplasm occurs in infants and young children (median age 8 months) and show a favorable prognosis. On gross examination, type I PPB presents as unilocular or more often multilocular cysts located at the periphery of the lung. On histological examination, the cysts are lined by respiratory epithelium with cuboidal or columnar ciliated cells. A proliferation of small, round to spindle shaped mesenchymal cells (blastema) is present beneath the epithelium. These malignant mesenchymal cells, diagnostic of type 1 PPB, may be localized to a limited focus, several foci or may grow in a diffuse band beneath the epithelium, resembling the cambium layer of a botryoid rhabdomyosarcoma. Scattered cells with prominent eosinophilic cytoplasm, consistent with rhabdomyoblastic differentiation and small nodules of immature cartilage may be observed in the wall of the cysts. Type II PPB are partly solid and partly cystic tumors. Microscopically the cysts are identical to those observed in type I PPB, but the tumor cells within the cysts proliferate creating a grossly visible thickening of the septa or formation of a solid mass. Type III PPB are entirely solid neoplasms. The median ages at diagnosis for types II and III PPB are 35 and 41 months, respectively. Children with type II or III PPB typically present with weight loss, fever, chest pain, shortness of breath, and opacity on chest radiograph. Pneumothorax can also occur in type II PPB. Microscopically, the solid areas of type II and III PPB are characterized histologically by a mixture of primitive blastematous and sarcomatous elements (Figure 2). The blastematous component is composed of small undifferentiated cells with scanty cytoplasm, round to ovoid nuclei and numerous mitoses. The mesenchymal component can show one or more sarcomatous patterns, including undifferentiated sarcoma, EMRS, spindle cell sarcoma and cartilage, usually with malignant features. Rhabdomyosarcomatous pattern is the most common and consists of a proliferation of polygonal or elongated rhabdomyoblasts with prominent eosinophilic cytoplasm occurring singly or in clusters and sheets [30]. Area of necrosis, hemorrhage and myxoid degeneration are variably present. In contrast to type I PPB, 75% of type II PPB and in 85% of type III PPB, show foci of anaplasia, especially in the undifferentiated and rhabdomiosarcomatous areas. Immunohistochemistry is not essential for the diagnosis but can be used to confirm the rhabdomyoblastic or cartilagineous differentiation. Type I PPB has no metastatic potential and a 5 years’ disease free survival of 80-90%. Type II or III PPB can present with metastasis to the brain, bone, local thoracic lymph nodes, and liver. These neoplasms have a poor prognosis with a 5 years’ survival rate of less than 50%, even after multimodality therapy [31].
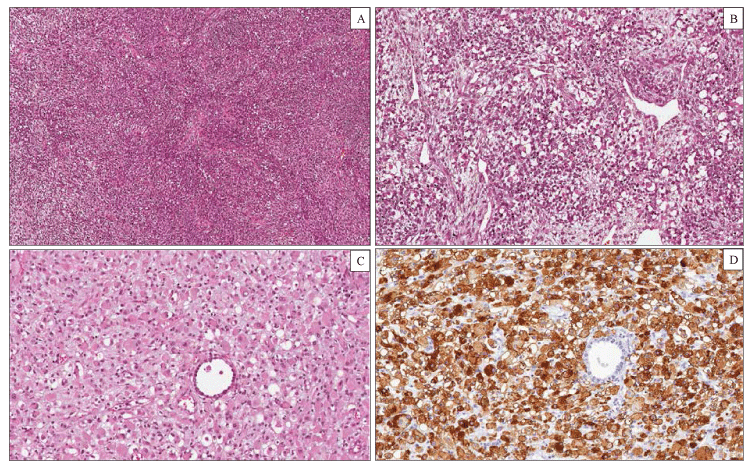
Figure 2: Pleuropulmonary Blastoma type III. Histological examination showing a solid tumor composed of a mixture of blastemal and mesenchymal neoplastic cells (hematoxylin-eosin). (A) Large areas of the tumor consisted of spindle cells arranged in intersecting fascicles with a brosarcoma-like growth pattern. (B) Other elds of the tumor showed a proliferation of blastemal cells and rhabdomyoblasts in a myxoid stroma. (C) Rhabdomyoblasts displayed an oval shape, eccentric nuclei and a densely eosinophilic cytoplasm. (D) Diuse cytoplasmic immunohistochemical staining for desmin in rhabdomyoblastic foci.
DISCUSSION
Rhabdomyoblasts are primitive mesenchymal cells showing variable degree of skeletal muscle differentiation. The detection of these cells on histological examination is considered one of the main parameters for the diagnosis of rhabdomyosarcoma. In this review, we emphasized that rhabdomyoblastic differentiation may be encountered in several pediatric neoplasms other than RMS, including nephroblastoma, pleuropulmonary blastoma, malignant triton tumour, infantile rhabdomyofibrosarcoma and malignant ectomesenchymoma. The awareness that rhabdomyoblastic and skeletal muscle differentiation are not limited to rhabdomyosarcoma, is crucial not only to correctly classify these neoplasms but also because the chemotherapy protocols used to treat patients with RMS may be different than those of other pediatric tumours in the differential diagnosis. Clinicopathological features as well as prognostic and treatment strategies for pediatric neoplasms showing rhabdomyoblastic differentiation are provided in table 1.
| Table 1: Clinical and prognostic features of Pediatric Soft Tissue neoplasms showing rhabdomyoblastic differentiation. | |||
| Age of presentation | Prognosis (5year survival rates) |
Treatment | |
| RMS | 0-20 years | Embryonal 66% Alveolar 54% Spindle cell/sclerosing 88% |
CT/ RT/WLE |
| MPNST | 28–36 years | 39% | WLE/ RT |
| MEM | 0-5 years | ND | WLE/ RT |
| WT | 0-15 anni | 90% | WLE/CT |
| PPB | 0-5 years | ND | WLE/ RT |
| IRMFS | 3 months to 3 years | ND | WLE/ CT |
| Abbreviations: WLE, wide local excision; CT, chemotherapy; RT, radiotherapy | |||
REFERENCES
- Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013; 20: 387-397. Ref.: https://goo.gl/sUAFuX
- Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006; 130: 1454-1465. Ref.: https://goo.gl/S8vwk7
- Miettinen M, Fetsch JF, Antonescu CR. Tumors with skeletal muscle differentiation. AFIP atlas of tumor pathology: tumors of the soft tissues. Silver Spring: ARP Press. 2014; 289-308.
- Woodruff JM, Perino G. Non-germ-cell or teratomatous malignant tumors showing additional rhabdomyoblastic differentiation, with emphasis on the malignant Triton tumor. Semin Diagn Pathol. 1994; 11: 69-81. Ref.: https://goo.gl/rg8lUB
- Fletcher CDM. Spindle cell/sclerosing rhabdomyosarcoma. World Health Organization (WHO) classification of tumours of soft tissue and bone. Vol 5. Fourth Edn. IARC press: France, Lyon, 2013; 468.
- Qualman SJ, Coffin CM, Newton WA, Hojo H, Triche TJ, et al. Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol. 1998; 1: 550-61. Ref.: https://goo.gl/PS6z00
- Cavazzana AO, Schmidt D, Ninfo V, Harms D, Tollot M, et al. Spindle cellrhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol. 1992; 16: 229-235. Ref.: https://goo.gl/UzGfAW
- Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002; 26: 1175-1183. Ref.: https://goo.gl/4ipwjA
- Rekhi B, Upadhyay P, Ramteke MP, Dutt A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod Pathol. 2016; 29: 1532-1540. Ref.: https://goo.gl/be0R7M
- Mungan S, Arslan S, Küçüktülü E, Ersöz Ş, Çobanoğlu B. Pleomorphic Rhabdomyosarcoma Arising from True Vocal Fold of Larynx: Report of a Rare Case and Literature Review. Case Rep Otolaryngol. 2016; 2016: 8135967. Ref.: https://goo.gl/6E9E7H
- Rudzinski ER, Anderson JR, Lyden ER, Bridge JA, Barr FG, et al. Myogenin, AP2β, NOS-1, and HMGA2 are surrogate markers of fusion status in rhabdomyosarcoma: a report from the soft tissue sarcoma committee of the children's oncology group. Am J Surg Pathol. 2014; 38: 654-659. Ref.: https://goo.gl/44GfNS
- Miki H, Kobayashi S, Kushida Y, Sasaki M, Haba R, et al. A case of infantile rhabdomyofibrosarcoma with immunohistochemical, electronmicroscopical, and genetic analyses. Hum Pathol. 1999; 30: 1519-1522. Ref.: https://goo.gl/XrNi6K
- Lundgren L, Angervall L, Stenman G, Kindblom LG. Infantile rhabdomyofibrosarcoma: a high-grade sarcoma distinguishable from infantile fibrosarcoma and rhabdomyosarcoma. Hum Pathol. 1993; 24: 785-795. Ref.: https://goo.gl/KmOrQD
- Stasik CJ, Tawfik O. Malignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation (malignant triton tumor). Arch Pathol Lab Med. 2006, 130: 1878-1881. Ref.: https://goo.gl/8BLODr
- Durbin AD, Ki DH, He S, Look AT. Malignant Peripheral Nerve Sheath Tumors. Adv Exp Med Biol. 2016; 916: 495-530. Ref.: https://goo.gl/n8eirv
- Masson P. Recklinghausen’s neurofibromatosis, sensory neuromas and motor neuromas. In: Libman Anniversary. Vol 2. New York, NY: International Press. 1932: 793-802.
- Woodruff JM, Perino G. Non-germ-cell or teratomatous malignant tumors showing additional rhabdomyoblastic differentiation, with emphasis on the malignant triton tumor. Semin Diagn Pathol. 1994; 11: 69-81. Ref.: https://goo.gl/rTiRHj
- Karcioglu Z, Someren A, Mathes SJ. Ectomesenchymoma. A malignant tumor of migratory neural crest (ectomesenchyme) remnants showing ganglionic, schwannian, melanocytic and rhabdomyoblastic differentiation. Cancer. 1977; 39: 2486-2496. Ref.: https://goo.gl/dR4vdr
- Huang SC, Alaggio R, Sung YS, Chen CL, Zhang L, et al. Frequent HRAS Mutations in Malignant Ectomesenchymoma: Overlapping Genetic Abnormalities with Embryonal Rhabdomyosarcoma. Am J Surg Pathol. 2016; 40: 876-885. Ref.: https://goo.gl/cbhfNV
- Chau YY, Hastie ND. The role of Wt1 in regulating mesenchyme in cancer, development, and tissue homeostasis. Trends Genet. 2012; 28: 515-524. Ref.: https://goo.gl/gBcxA1
- Salvatorelli L, Parenti R, Leone G, Musumeci G, Vasquez E, et al. Wilms tumor 1 (WT1) protein: Diagnostic utility in pediatric tumors. Acta Histochem. 2015; 117: 367-378. Ref.: https://goo.gl/Cy7A8U
- Tu BW, Ye WJ, Li YH. Botryoid Wilms' tumor: report of two cases. World J Pediatr. 2011; 7: 274-276. Ref.: https://goo.gl/enFS3P
- Carpentieri DF, Nichols K, Chou PM, Matthews M, Pawel B, et al. The expression of WT1 in the differentiation of rhabdomyosarcoma from other pediatric small round blue cell tumors. Mod Pathol. 2002; 15: 1080-1086. Ref.: https://goo.gl/JH5dM6
- Pollono D, Drut R, Tomarchio S, Fontana A, Ibañez O. Fetal rhabdomyomatous nephroblastoma: report of 14 cases confirming chemotherapy resistance. J Pediatr Hematol Oncol. 2003; 25: 640-643. Ref.: https://goo.gl/NKaglc
- Irtan S, Ehrlich PF, Pritchard-Jones K. Wilms tumor: "State-of-the-art" update, 2016. Semin Pediatr Surg. 2016; 25: 250-256. Ref.: https://goo.gl/nKgxCA
- Cone EB, Dalton SS, Van Noord M, Tracy ET, Rice HE, et al. Biomarkers for Wilms Tumor: A Systematic Review. J Urol. 2016; 196: 1530-1535. Ref.: https://goo.gl/8XxcXX
- Fosdal MB. Pleuropulmonary blastoma. J Pediatr Oncol Nurs. 2008; 25: 295-302. Ref.: https://goo.gl/AFy4I5
- Mehraein Y, Schmid I, Eggert M, Kohlhase J, Steinlein OK. DICER1 syndrome can mimic different genetic tumor predispositions. Cancer Lett. 2016; 370: 275-278. Ref.: https://goo.gl/rFlSyv
- Hill DA, Jarzembowski JA, Priest JR, Williams G, Schoettler P, et al. Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the international pleuropulmonary blastoma registry. Am J Surg Pathol. 2008; 32: 282-295. Ref.: https://goo.gl/W3ANsO
- Sciot R, Dal Cin P, Brock P, Moerman P, Van Damme B, et al. Pleuropulmonary blastoma (pulmonary blastoma of childhood): genetic link with other embryonal malignancies? Histopathology. 1994; 24: 559-563. Ref.: https://goo.gl/XjHWbI
- Indolfi P, Bisogno G, Casale F, Cecchetto G, De Salvo G, et al. Prognostic factors in pleuro-pulmonary blastoma. Pediatr Blood Cancer. 2007; 48: 318-23. Ref.: https://goo.gl/1HwiOh